说明:以二维InSe原胞为例,QE版本:,赝势:SSSP。
1.结构优化
通过Materialsstudio2018的建模功能切出二维InSe原胞,如图所示,并导出为格式。

二维InSe原胞
2、QE输入文件结构
QE的结构优化主要使用的是模块,该模块包含非常多的输入参数,建议在计算过程中逐步学习,无需一次性学完所有命令,即使学完也是徒劳,因为几乎无法记住。
结构优化在QE中是比较诟病的一个地方,优化速度非常慢,对于大体系实现结构优化是非常慢的,如果计算的体系包含较多的原子,建议使用VASP。QE结合EPW计算材料的与声子有关的性质比较合适,且这类计算一般不需要建立超胞,能够达到计算目的。总之,根据计算的任务和目的合理选择第一性原理计算软件是比较明智的。
2.1模块输入文件结构
CONTROL/SYSTEM/ELECTRONS/[IONS/][CELL/]ATOMIC_SPECIESXMass_XPseudoPot_XYMass_YPseudoPot_YZMass_ZPseudoPot_ZATOMIC_POSITIONS{alat|bohr|crystal|angstrom|crystal_sg}{if_pos(1)if_pos(2)if_pos(3)}K_POINTS{tpiba|automatic|crystal|gamma|tpiba_b|crystal_b|tpiba_c|crystal_c}if(gamma)nothingtoreadif(automatic)nk1,nk2,nk3,k1,k2,k3if(notautomatic)nksxk_x,xk_y,xk_z,wk[CELL_PARAMETERS{alat|bohr|angstrom}v1(1)v1(2)v1(3)v2(1)v2(2)v2(3)v3(1)v3(2)v3(3)][OCCUPATIONSf_inp1(1)f_inp1(2)f_inp1(3)f_inp1(10)f_inp1(11)f_inp1(12)f_inp1(nbnd)[f_inp2(1)f_inp2(2)f_inp2(3)f_inp2(10)f_inp2(11)f_inp2(12)f_inp2(nbnd)]][CONSTRAINTSnconstr{constr_tol}constr_type(.)constr(1,.)constr(2,.)[constr(3,.)constr(4,.)]{constr_target(.)}][ATOMIC_FORCESlabel_1Fx(1)Fy(1)Fz(1)..label_nFx(n)Fy(n)Fz(n)]2.2二维InSe原胞结构优化输入文件
CONTROLcalculation='vc-relax',!vc-relax|relaxrestart_mode='from_scratch',!normalusedprefix='InSe',!prepedtoinput/outputfilenamespseudo_dir='../pseudo/',!directorycontainingpseudopotentialfilesoutdir='../tmp/',!input,temporary,outputfilesarefoundinthisdirectoryforc_conv_thr=1d-5,!forcesconvergencethreshold1d-03/SYSTEMibrav=0!Bravais-latticeindexnat=4!numberofatomsintheunitcellntyp=2!numberoftypesofatomsintheunitcellecutwfc=60.0,!kineticenergycutoff(Ry)forwavefunctionsecutrho=720.0,!Kineticenergycutoff(Ry)forchargedensity/ELECTRONSmixing_beta=0.7,!mixingfactorforself-consistencyconv_thr=1d-8,!Convergencethresholdforselfconsistency1d-6/IONSion_dynamics='bfgs',!Specifythetypeofionicdynamics/CELLcell_dynamics='bfgs',!Specifythetypeofdynamicsforthecellpress=0.0,!press_conv_thr=0.5,!Convergencethresholdonthepressureforvariablecellcell_dofree=2Dxy,!for2Dmaterials/CELL_PARAMETERS{angstrom}!specifythestructure4.08360004430.00000000000.0000000000-2.04180002213.53650137720.00000000000.00000000000.000000000025.3775005341ATOMIC_SPECIES!__pbe_ATOMIC_POSITIONS{crystal}K_POINTSautomatic121210002.3结构优化的经验总结
模块用于结构优化时,对于体系的对称性非常敏感,也就是QE自身不会寻找体系的对称性,只能依靠手动输入体系的对称性,这点与VASP相比是非常欠缺的,因为VASP是可以自己寻找对称性的。对于二维InSe原胞,有对称性与无对称性的结构优化使用时间相差了近5倍。
查看优化所用时间
|tail-n1
输入对称性
PWSCF:4
没有输入对称性
PWSCF:21
对于离子的受力,一般使用准牛顿迭代算法
ion_dynamics='bfgs'
pwscf模块做结构优化时,对于二维材料无需重新编译软件,即可做到不优化真空层,这点比VASP方便,具体设置如下
cell_dofreeCHARACTERDefault:'all'Selectwhichofthecellparametersshouldbemoved:'all':allaxisandanglesaremoved'ibrav':allaxisandanglesaremoved,butthelatticebutberepresentablewiththeinitialibravchoice'x':onlythexcomponentofaxis1(v1_x)ismoved'y':onlytheycomponentofaxis2(v2_y)ismoved'z':onlythezcomponentofaxis3(v3_z)ismoved'xy':onlyv1_xandv2_yaremoved'xz':onlyv1_xandv3_zaremoved'yz':onlyv2_yandv3_zaremoved'xyz':onlyv1_x,v2_y,v3_zaremoved'shape':allaxisandangles,keepingthevolumefixed'volume':thevolumechanges,keepingallanglesfixed((1)changes)'2Dxy':onlyxandycomponentsareallowedtochange'2Dshape':asabove,keepingtheareainxyplanefixed'epitaxial_ab':fixaxis1and2whileallowingaxis3tomove'epitaxial_ac':fixaxis1and3whileallowingaxis2tomove'epitaxial_bc':fixaxis2and3whileallowingaxis1tomoveBEWARE:ifaxisarenotorthogonal,,editsubroutineinit_dofreeinfileModules/cell_
3、结构自洽计算
3.1输入文件
CONTROLcalculation='scf',restart_mode='from_scratch',prefix='InSe',pseudo_dir='../pseudo/',outdir='../tmp/',/SYSTEMibrav=0nat=4ntyp=2ecutwfc=60.0,ecutrho=480.0,/ELECTRONSmixing_beta=0.7,conv_thr=1d-8,/CELL_PARAMETERS{angstrom}4.0819105670.0000000000.000000000-2.0409552833.5350382470.0000000000.0000000000.00000000025.377500534ATOMIC_SPECIES__pbe_ATOMIC_POSITIONS{crystal}K_POINTSautomatic12121000
$$ecutwfc
grep-e'kinetic-energycutoff'-e!$ecutwfc|\awk'/kinetic/{ecutwfc=$(NF-1)}/!/{printecutwfc,$(NF-1)}'_vs_ecutwfc
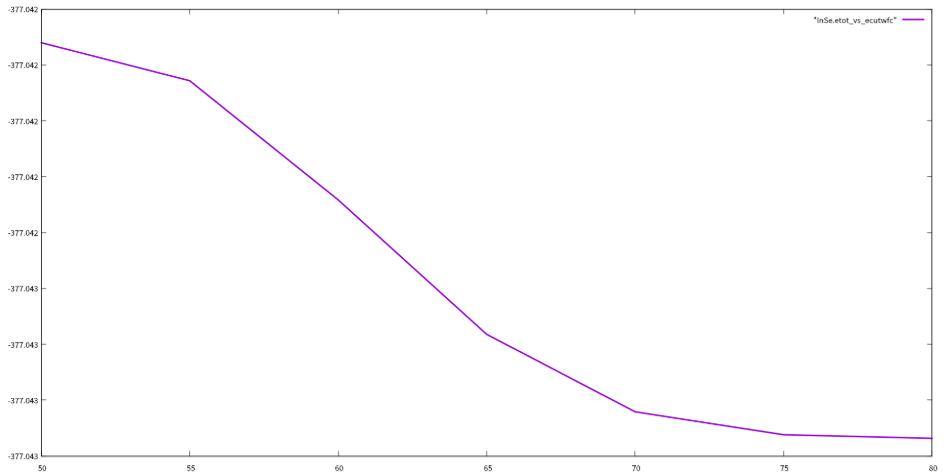
done3.2ecutwfc测试
forecutwfcin50556065707580;do
self-consistentcalculation$kpointsEOFCONTROLcalculation='scf',restart_mode='from_scratch',prefix='InSe',pseudo_dir='../pseudo/',outdir='../test_tmp/',/SYSTEMibrav=0nat=4ntyp=2ecutwfc=70,ecutrho=840,/ELECTRONSmixing_beta=0.7,conv_thr=1d-8,/CELL_PARAMETERS{angstrom}4.0819105670.0000000000.000000000-2.0409552833.5350382470.0000000000.0000000000.00000000025.377500534ATOMIC_SPECIES__pbe_ATOMIC_POSITIONS{crystal}K_POINTSautomatic$kpoints$kpoints1000EOF
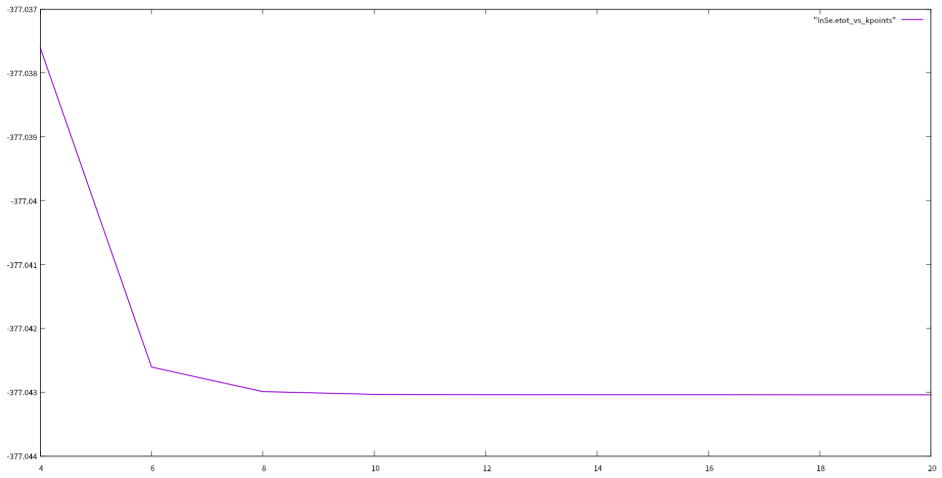
$$kpoints
echo-e"$kpoints\c"_vs_kpointsgrep!$kpoints|\awk'/!/{print$(NF-1)}'_vs_kpoints
done
测试结果:
kpoints
3.4结构自洽计算总结
通过对ecutwfc和kpoints的测试,我们选定ecutwfc的值为80Ry,kpoints的值为881,用这个值返回去重新优化结构和自洽,然后开始计算下面的性质。
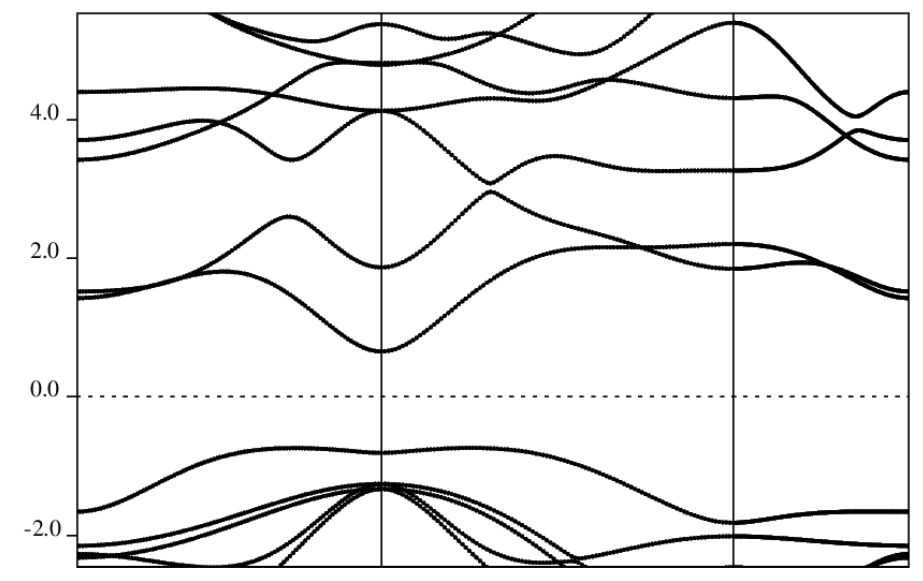
4、能带计算
4.1输入文件
CONTROLcalculation='bands',restart_mode='from_scratch',prefix='InSe',pseudo_dir='../pseudo/',outdir='../tmp/',verbosity='high',/SYSTEMibrav=0nat=4ntyp=2ecutwfc=80.0,ecutrho=960.0,nbnd=26,/ELECTRONSmixing_beta=0.7,conv_thr=1d-8,/CELL_PARAMETERS{angstrom}4.085963663-0.0000000000.000000000-2.0429818323.5385483310.0000000000.0000000000.00000000025.377500534ATOMIC_SPECIES__pbe_ATOMIC_POSITIONS{crystal}K_POINTScrystal_b40.50.00.0100!M0.00.00.0100!G0.333330.333330.0100!K0.50.00.0100!M4.2后处理文件文件
bandsprefix='InSe',outdir='../tmp/',filband='',lsym=.true./
4.3画能带图的后处理程序
Reading26bandsat301k-pointsfilewithrepresentationsnotcompatiblewithbandsRange:-16.45005.4820eVEmin,Emax-44high-symmetrypoint:0.50000.28870.0000high-symmetrypoint:0.00000.00000.0000high-symmetrypoint:0.33330.57730.0000high-symmetrypoint:0.50000.28870.0000outputfile(gnuplot/xmgr)bandsingnuplot/outputfile(ps)deltaE,referenceE(fortics)2-1.5444
说明:QE自带的画图程序画出的能带图几乎无法直接用于文章中,因此这步处理意义不大。可以直接使用计算出的文件,通过origin等专业的绘图软件绘制能带图。
能带图计算结果:
2D_InSe-band_qe
链接:
5、其它QE入门教程友情链接
(1)
(2)
(3)





